

1. Genealogías
1.1. Símbolos usados
1.2. Símbolos usados
1.2.1. Probando o propositus hace referencia al sujeto de estudio
1.3. Se utilizan para
1.3.1. Interpretar relaciones de parentesco entre los individuos
1.3.2. Determinar el modo de herencia del carácter en estudio
1.3.2.1. Recesivo o dominante
1.3.2.2. Autosómico o ligado al sexo
1.3.3. Analizar probabilidades de que una persona sea portadora o tenga un hijo que exprese el caracter
1.4. En humanos hay 44 autosomas más un par sexual (XX o XY)
2. Enfermedades Autosómicas Recesivas
2.1. Albinismo
2.1.1. OCA1 en el cromosoma 11
2.1.1.1. Albinismo ocular tipo 1
2.1.1.2. Dañada la expresión de la melanina en la piel y en los pigmentos del ojo
2.1.2. Mecanismo
2.1.2.1. Tirosinasa reconoce a tirosina y la transforma en DOPA
2.1.2.2. Luego, toma a DOPA y la convierte en DOPAquinona
2.1.2.3. DOPAquinona sufre reacciones metabólicas conducentes a la síntesis de
2.1.2.3.1. Eumelaninas (negras o marrones)
2.1.2.3.2. Feomelaninas (amarillas o rojas)
2.1.2.3.3. Que dan la tonalidad de piel y pelo de las distintas "razas" humanas
2.1.2.4. Tirosina mutada en albinismo
2.1.2.4.1. Albinos no tienen melaninas
2.1.3. Genealogía para albinismo
2.1.3.1. Por qué no salen marcados los heterocigotos para el gen?
2.1.3.2. Ejercicio: Si I1 y I2 portan el alelo recesivo, ¿cuáles serán los probables genotipos y fenotipos de la descendencia? NOTA: se cumple le ley de Mendel, no se cumplen las proporciones esperadas
2.1.4. Características
2.1.4.1. Aparece generalmente en la descendencia de padres no afectados
2.1.4.2. Se afectan por igual hombres y mujeres
2.1.4.2.1. Característica de la herencia mendeliana simple de un gen autosómico
2.1.4.3. Puede saltar varias generaciones sin manifestarse
2.1.4.4. Es más común entre progenitores con algún rgado de consanguinidad
2.2. Fenilcetonuria
2.2.1. A muchos genotipos se les puede atenuar el fenotipo
2.2.1.1. Personas que tengan la mutación pueden no presentar síntomas clínicos
2.2.1.2. Intervención dietética baja en proteínas hace que enfermos no padezcan el retraso mental o éste se aminore
2.2.2. Mutación hace que no haya fenilalanina hidroxilasa
2.2.2.1. No se metaboliza la fenilalanina (presente en proteínas)
2.2.2.2. Al consumir proteínas, aumenta fenilalanina en circulación y aumento deteriora el sistema nervioso, causando retardo mental
2.3. Fibrosis Quística
2.3.1. Mutación ocurre en un canal de cloruro
2.3.2. Secreciones más densas y abundantes a nivel pulmonar
2.3.2.1. Favorece las infecciones
2.3.3. Puede tener muerte temprana
3. Enfermedades Autosómicas Dominantes
3.1. Ejemplo de genealogía
3.2. Características
3.2.1. Alelo recesivo es el normal, alelo dominante genera la patología
3.2.2. Fenotipo patológico aparece en cada generación
3.2.3. Cada individuo afectado tiene al menos un progenitor enfermo
3.2.4. Se afectan igual hombres y ujeres
3.2.5. También pueden surgir debido a mutaciones de novo que ocurren en la gametogénesis
3.3. Ejemplos
3.3.1. Pseudoacondroplasia
3.3.1.1. Mutación en Gen COMP (19p13.1)
3.3.1.1.1. Codifica para la proteína oligomérica de la matriz del cartílago
3.3.1.2. Características
3.3.1.2.1. Retraso en el crecimiento
3.3.1.2.2. Piernas arqueadas
3.3.1.2.3. Escoliosis
3.3.1.3. Caso: Familia Ovitz
3.3.1.3.1. Padecían pseudoacondroplasia
3.3.1.3.2. El padre de la familia estaba afectado
3.3.1.3.3. Los descendientes tuvieron talla normal
3.3.2. Enfermedad de Huntington
3.3.3. Polidactilia
3.3.4. Poliquistosis renal
3.3.4.1. 1/1000 (incidencia alta)
3.3.4.2. Mutación en una proteína de membrana
3.3.4.2.1. Gen mutado PKD1: 90% de los casos
3.3.4.2.2. Gen mutado PKD2: 10% de los casos
3.3.4.2.3. Codifican para la policistina
3.3.4.3. Hay heterogeneicidad del fenotipo
3.3.4.3.1. Dependiendo de la alimentación (si comen mucha proteína) necesitarán diálisis o no
3.3.4.4. En condiciones normales
3.3.4.4.1. Policistina-1 interactúa en un complejo multiproteico con integrinas y proteínas de adhesión focal, como la quinasa de adhesión focal (FAK)
3.3.5. Poliquistosis hepática
4. Interacción génica
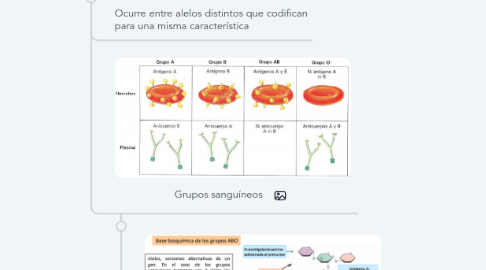
4.1. Ocurre entre alelos distintos que codifican para una misma característica
4.2. Grupos sanguíneos
4.2.1. Base bioquímica
4.2.1.1. Molecula precursora es modificada a Molécula (H) a través de una UDP-Fuc
4.2.1.1.1. Transferasa H (UDP-Fuc -> UDP)
4.2.1.1.2. Molécula H: N-acetilglucosamina + Fucosa + Galactosa
4.2.2. Cada alelo modifica, a través de transferasas distintas, la galactosa terminal de la molécula H (estos alelos están en el cromosoma 9)
4.2.2.1. Transferasa A (UDP-GalNAc -> UDP)
4.2.2.1.1. Le agrega N-acetilgalactosamina a la sustancia precursora
4.2.2.1.2. Antígeno A + Fucosa + GalNac
4.2.2.1.3. Alelo A
4.2.2.2. Transferasa B (UDP-Gal -> UDP)
4.2.2.2.1. Le agrega Galactosa a la sustancia precursora
4.2.2.2.2. Antígeno B + Fucosa + Galactosa
4.2.2.2.3. Alelo B
4.2.2.3. Alelo O no codifica una transferasa funcional
4.2.2.3.1. Antígeno O (H) + Fucosa
4.2.2.3.2. No reacciona con anticuerpos anti-A ni anti-B
4.2.2.3.3. Plasma de alguien con grupo O tiene anticuerpos anti-a y anti-b
4.2.3. Codominancia de alelos A y B sobre alelo i
4.2.4. Desarrollo de los antígenos del sistema ABO
4.2.5. Fenotipo Bombay (Oh)
4.2.5.1. Genotipo hh
4.2.5.2. No expresa la molécula H
4.2.5.2.1. Ya que se produce una transferasa no funcional incapaz de modificar la molécula precursora
4.2.5.2.2. No tienen el alelo para la transferasa A o B en qué actuar
4.2.5.3. Solo recibe sangre de su mismo tipo
4.2.5.4. Se enmascaran los fenotipos AB, A, B y O
4.2.5.5. Epistasis: Dos genes que trabajan en la misma ruta
4.2.5.5.1. Dos genes trabajando para determinar el tipo de sangre: los alelos para transferasas A y B y el alelo H
4.2.5.6. Epistasis Recesiva: Un alelo recesivo enmascara la expresión de otro gen (En este caso hh)
4.2.5.6.1. Otro ejemplo de epistasis recesiva: labradores
4.2.5.7. Genotipo homocigoto recesivo (hh) se expresa, no se puede expersar el otro gen tampoco (transferasa A o B)
5. Herencia ligada al sexo
5.1. En el cromosoma Y se encuentra el gen SRY (factor determinante de los testículos)
5.2. Genes DAZ: ausentes en azoospermia
5.2.1. Ausencia total o casi total de espermios en el semen
5.3. Aspectos de los cromosomas sexuales en humanos:
5.3.1. Cromosoma X esencial para desarrollo humano
5.3.2. Copias adicionales de cromosmoa X afecta la fertilidad y el desarrollo mental
5.3.3. SRY mapea en el cromosoma Y
5.3.4. Un cromosoma X mutante portador de un gen SRY determina el sexo masculino en un XX
5.3.4.1. Esto puede ocurrir ya que gen SRY está muy cerca de la zona pseudoautosómica del cromosoma Y y se puede haber recombinado con el cromosoma X
5.3.4.2. Individuos no tendrán genes DAZ y no producirán espermios
5.4. Compensación de la dosis génica
5.4.1. Mamiferos: Corpúsculo de Barr
5.4.1.1. Cromatina muy condensada perteneciente al cromosoma X que da cuenta del cromosoma inactivado
5.4.1.1.1. Es visible al microscopio previa tinción
5.4.2. La inactivación de un cromosoma X ocurre al azar en el desarrollo embrionario de la mujer
5.4.3. Produce individuos mosaico
5.4.4. X - 1 de la célula se inactivan al azar dando lugar al Corpúsculo de Barr
5.4.4.1. Esta inactivación es parcial
5.4.4.1.1. Los genes de la zona pseudoautosómica escapan de la inactivación
5.4.5. Inactivación al azar de un cromosoma X en mujeres
5.4.5.1. Actúa Xist: RNA largo no codificante
5.4.5.1.1. Transcrito del gen Xist, presente en el cormosoma que se va a inactivar
6. Genoma mitocondrial
6.1. Función de la mitocondria
6.1.1. Abundantes en los órganos y tejidos del organismo con mayores requerimientos energéticos.
6.1.1.1. 80% del volumen de células fotorreceptoras oculares corresponde a mitocondrias.
6.1.1.2. 40% del volumen de células musculares cardíacas corresponde a mitocondrias.
6.2. Genoma mitocondrial humano
6.2.1. Composición
6.2.1.1. 16,569 pares de bases
6.2.1.2. 37 genes
6.2.1.3. 2 rRNA
6.2.1.4. 22 tRNA
6.2.1.5. 13 polipéptidos
6.2.2. Características
6.2.2.1. DNA circular doble hebra
6.2.2.2. Super-enrollado
6.2.2.3. No posee histonas
6.2.2.4. Cada mitocondria posee múltiples copias (2-10 por organelo)
6.2.2.5. No hay intrones
6.2.2.6. Se hereda por vía materna
6.2.3. Variaciones en el código genético
6.2.3.1. UGA (codón stop) se traduce como triptófano
6.2.3.2. AGG (arginina) se lee como codón de detención de síntesis de proteínas
6.2.4. Alta tasa de mutuación
6.2.4.1. 10 veces superior al DNA nuclear
6.2.4.2. Dos individuos al azar pueden presentar diferencias de 50-70 nucleótidos
6.2.5. Homoplasmia
6.2.5.1. Todas las células de un individuo normal tienen moléculas idénticas de mtDNA
6.2.6. Heteroplasmia
6.2.6.1. Ocurre cuando en una célula aparecen mitocondrias conteniendo mtDNA normal y mutado.
6.2.6.2. El fenotipo de una célula en heteroplasmia dependerá del porcentaje de ADN dañado que exista en la célula.
6.2.6.2.1. Si el número de moléculas de ADNmt mutadas es pequeño, el ADNmt normal es capaz de mantener la función mitocondrial.
6.2.6.3. Efecto umbral
6.2.6.3.1. Número de copias de ADNmt mutado sobrepasa un umbral determinado, la producción de ATP puede llegar a estar por debajo de los mínimos necesarios para el funcionamiento de los tejidos (patología mitocondrial)
6.2.6.3.2. Los distintos tejidos del cuerpo humano pueden variar en el grado de homoplasmia o heteroplasmia
6.2.7. Segregación mitótica
6.2.7.1. Las mitocondrias (y las moléculas de ADNmt) se distribuyen al azar entre las células hijas.
6.2.7.1.1. Gracias a esto, un hijo puede salir tremendamente enfermo o sano, ya que podrían segregarse un gran número de mitocondrias mutadas a un gameto y ni una mitocondria mutada a otro gameto
6.2.8. Genealogía de una enfermedad mitocondrial
6.2.8.1. Se observa que hay cuatro mujeres portadoras asintomáticas -debido a los distintos grados de heteroplasmia-
6.2.8.2. Con frecuencia las mujeres que transmiten la enfermedad no llegan a desarrollar el cuadro clínico completo
7. Otros datos
7.1. Dominancia y Recesividad indican cómo actúan los alelos. Eso no significa que un alelo dominante sea abundante en la población.
7.2. Penetrancia
7.2.1. Proporción de individuos de una población, con un mismo genotipo (con un alelo mutado), que expresan el fenotipo patológico
7.2.2. Heterocigotos afectados/Heterocigotos totales
7.3. Penetrancia incompleta (Penetrancia menor a 1)
7.3.1. No todos los individuos con el genotipo expresarán el correspondiente fenotipo, debido a
7.3.1.1. Influencia del ambiente
7.3.1.2. Influencia de otros genes, ejemplo, genes epistáticos, que previenen la expresión de lfenotipo estándar
7.3.2. Ejemplo
7.3.2.1. Polidactilia causada por un alelo dominante P_
7.3.2.2. Algunas personas con el gen dominante tienen manos y pies con el número normal de dedos
7.4. Expresividad Variable
7.4.1. Variación en la expresión de un alelo
7.4.2. Se mide la intensidad del fenotipo
7.4.3. Ejemplo: polidactilia